本指导原则旨在指导注册申请人对体外除颤产品(简称“产品”)注册申报资料的准备及撰写,同时也为技术审评部门审评注册申报资料提供参考。
本指导原则是对产品的一般性要求,注册申请人应依据其具体特性确定其中内容是否适用,若不适用,需具体阐述理由及相应的科学依据,并依据其具体特性对注册申报资料的内容进行充实和细化。
本指导原则是供注册申请人和审查人员使用的指导文件,不涉及注册审批等行政事项,亦不作为法规强制执行,如有能够满足法规要求的其他方法,也可以采用,但应提供详细的研究资料和验证资料,应在遵循相关法规的前提下使用本指导原则。
本指导原则是在现行法规、标准体系及当前认知水平下制定的,随着法规、标准体系的不断完善和科学技术的不断发展,本指导原则相关内容也将适时进行调整。
一、范围
本指导原则适用于体外除颤产品,可进行体外除颤治疗(简称“除颤”)。
按照《医疗器械分类目录》,体外除颤产品的管理类别为Ⅲ类,分类编码为6821,是用于心脏的治疗、急救装置。
本指导原则所述的产品包含进行除颤的各种设备或系统。产品既可以是独立设备,也可集成在多参数模块的设备或系统中,也可以是穿戴式的;产品可以全部由执业医生控制和操作来进行手动除颤,或者只需急救者按确认除颤键来进行半自动除颤,或者无须任何人为干预来进行自动除颤。
本指导原则对于产品的预期使用环境不做限制,例如,手动体外除颤器和频繁使用的半自动体外除颤器可在医疗机构中和院前的急救环境中使用;非频繁使用的半自动体外除颤器和自动体外除颤器可在机场、生活社区、学校、办公场所等公共区域使用;穿戴式的自动体外除颤器在患者的日常生活中使用。
本指导原则不适用于植入式除颤产品。
二、医疗器械安全有效基本要求清单
依据《医疗器械注册申报资料要求和批准证明文件格式》(国家食品药品监督管理总局2014年第43号公告)附件8,注册申请人应提供《医疗器械安全有效基本要求清单》,着重明确下述项目的适用性,说明产品符合适用要求所采用的方法,提供证明其符合性的文件:
1.B5项环境特征;
2.B6项有诊断或测量功能的医疗器械产品;
3.B8项含软件的医疗器械和独立医疗器械软件;
4.B9项有源医疗器械和与其连接的器械;
5.B11项提供患者能量或物质而产生风险的防护;
6.B12项对非专业用户使用风险的防护。
对于包含在产品注册申报资料中的文件,应当说明其在申报资料中的具体位置;对于未包含在产品注册申报资料中的文件,应当注明该证据文件名称及其在质量管理体系文件中的编号备查。
三、综述资料
(一)概述
按照功能和预期用途,体外除颤分为手动体外除颤、半自动体外除颤、自动体外除颤。手动体外除颤分为胸外除颤和胸内除颤。
产品通用名称由核心词和特征词组成。注册申请人应按照产品的预期用途来确定产品名称的核心词,按照产品的使用部位和技术特点来确定产品名称的特征词,例如,手动体外除颤器、半自动体外除颤器、自动体外除颤器、体外除颤监护仪、体外除颤监护系统等。
(二) 产品描述
注册申请人应提供体外除颤产品的下述描述信息:
1.除颤的临床机理和需求、工作原理
注册申请人应描述人体在发生心律失常时的临床表现,应明确手动体外除颤、半自动体外除颤、自动体外除颤的分析和治疗机理、临床需求以及体外除颤产品的工作原理。
2.结构组成
注册申请人应明确产品组成的信息,包含但不限于:主机、部件和附件的名称、型号和法定制造商名称。这些信息同时应以注册申请表附页形式提供,例如,表1。
表 1
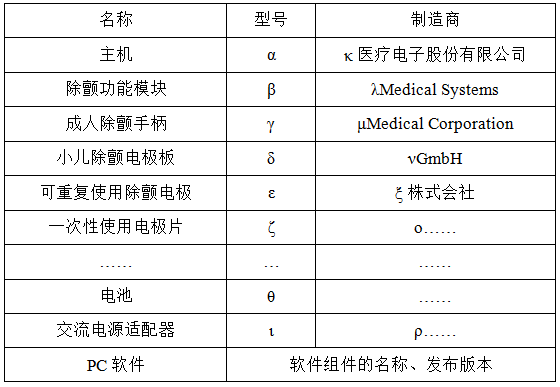
基于上述产品组成信息,制造商应提供各部分的示意图或者彩色图片,描述与产品配合使用的附件、主要功能及其组成部件(关键组件和软件)的功能,以及区别于其他同类产品的特征等内容。
(三) 型号规格
对于存在多种型号的体外除颤产品,应当明确各型号的区别。应当采用对比表及带有说明性文字的图片、图表,对于各种型号的结构、组成、配置和除颤治疗模式加以描述。
注册申请人应明确主机、部件和附件的规格,如物理规格、显示规格、有线/无线通信协议、电源/电池规格、打印规格等。
1.物理规格:物理尺寸、重量,等等。
2.显示规格:显示屏类型、尺寸。
3.有线/无线通信协议
a)硬件接口:接口的名称、机械和电气协议,例如,RS-232或RS-485串口,并口,USB 1.0、USB 2.0、USB 3.0,IEEE802.3协议的标准以太网口、快速以太网口和10G以太网口,SD/CF卡插口,等等。
b)无线接口:接口的名称及协议,例如,红外接口,802.11a/b/g/n协议的WIFI网络,IEEE 802.15.1协议的蓝牙(版本1.0—4.2),等等。
c)打印规格:打印机类型,例如,热敏型等;打印分辨率:垂直分辨率、水平分辨率等;纸张宽度、打印宽度;打印波形数量等;打印速度,例如,12.5、25mm/s。
4.附件的规格
a)附件的结构示意图/彩色图片、附件的各部分原材料。
b)附件的物理尺寸和面积、导线的物理尺寸等。
c)对于灭菌包装附件,应明确灭菌方式和灭菌有效期。
(四) 包装说明
注册申请人应提供体外除颤产品包装的信息及彩色图片,以及与该产品一起销售的配件包装情况及彩色图片。
(五) 适用范围和禁忌症
体外除颤产品是生命支持设备或系统,预期用于患者急救,例如:
1.手动体外除颤可终止心室颤动或室性心动过速症状,预期治疗新生儿、小儿和成人患者,应由经急救培训且接受设备操作培训的医护人员使用,可用于医院、临床机构、救护车、急救现场或者患者转运过程中。
2.半自动体外除颤可用于疑似心脏骤停患者,预期治疗新生儿、小儿和成人患者,应由经急救培训的人员或在急救中心调度人员指导下使用。
3.自动体外除颤可持续测量和分析患者的心电,如果患者出现危及生命的心室颤动或室性心动过速症状,该产品可自动进行体外除颤治疗。
某些自动体外除颤器可由经急救培训的人员使用。某些自动体外除颤器预期用于有心脏骤停风险、不适宜植入或拒绝使用植入式除颤器的成人患者。
注册申请人应当明确说明产品不适宜应用的某些疾病、症状、情况或特定的人群(如新生儿、小儿、老年人、孕妇及哺乳期妇女、肝肾功能不全者),例如:
1.半自动体外除颤器禁用于有脉搏、有呼吸、有意识的患者。
2.某些半自动体外除颤器禁用于年龄小于8岁或体重低于25千克的患者,某些半自动体外除颤器禁用于新生儿患者,等等。
(六) 同类产品或前代产品的信息
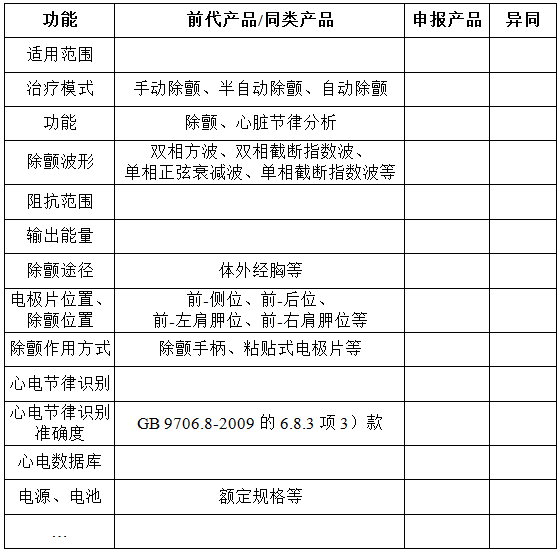
参考的同类产品或前代产品应当提供同类产品 ( 国内外已上市 ) 或前代产品 ( 如有) 的信息,阐述申请注册产品的研发背景和目的。对于同类产品,应当说明选择其作为研发参考的原因。同时列表比较说明申请注册产品与参考产品(同类产品或前代产品)的异同,例如,表2。
(七) 其他需说明的内容
对于已获得批准的部件或配合使用的附件,应当提供批准文号和批准文件复印件;预期与其他医疗器械或通用产品组合使用的应当提供说明;应当说明系统各组合医疗器械间存在的物理、电气等连接方式。
表 2
四、研究资料
(一)产品性能研究
注册申请人应当提供产品技术要求的研究和编制说明 。体外除颤产品的现行有效标准 如下:
GB 9706.1-2007 医用电气设备第1部分:安全通用要求;
GB 9706.8-2009 医用电气设备第2-4部分:心脏除颤器安全专用要求;
GB 9706.15-2008 医用电气设备第1-1部分:通用安全要求并列标准:医用电气系统安全要求(如适用);
YY 0505-2012医用电气设备第1-2部分 : 安全通用要求并列标准 :电磁兼容要求和 试验;
YY 0709-2009 医用电气设备第1-8部分:安全通用要求并列标准:通用要求,医用电气设备和医用电气系统中报警系统的测试和指南(如适用);
YY/T 0196-2005 一次性使用心电电极(如适用)。
注册申请人应当提供产品性能研究资料,包括性能指标、质量控制相关指标的确定依据,所采用的标准或方法、采用的原因及理论基础。
1.关键技术特点及其性能指标
注册申请人应明确产品的治疗模式,例如,手动模式、半自动模式、自动模式等。
注册申请人应明确各治疗模式下的功能和性能指标,例如,除颤波形、能量级别和精度、病人阻抗测量、心电波形和节律识别、充电/放电、电源等方面的功能和性能指标。
a)在所有的治疗模式下,注册申请人应明确除颤波形及其详细参数,例如,双相波(例如,双相方波RLB、双相截断指数波BTE,等等)、单相波(例如,单相正弦衰减波MDS、单相截断指数波MTE,等等)或其他波形,并且至少明确下述性能指标:
双相除颤波形的详细参数,例如:所有能量级别和各级别的输出能量精度、放电时间常数、第一相时间、第一相峰值电流、第一相平均电流、第一相电流平均斜率、两相间隔时间、第二相时间、第二相峰值电流、第二相平均电流、第二相电流平均斜率,等等。
单相除颤波形的详细参数,例如,所有能量级别和各级别的输出能量精度、放电时间常数、治疗时间、峰值电流、平均电流、波形斜率,等等。
按照电压或者电流—时间关系,注册申请人应绘制除颤波形释放的脉冲图,并针对波形特点提供波形详细参数,例如,图1和图2。
b)在所有的治疗模式下,注册申请人应提供阻抗测量的方法,应明确除颤波形对应的病人阻抗测量范围和精确度。
c)对于半自动和自动模式,注册申请人应描述心脏节律识别技术,陈述算法名称、算法原理和实现方式,明确心脏节律识别的性能指标,例如,分析时间、敏感度、特异性、真实预报价值和假阳性率等。
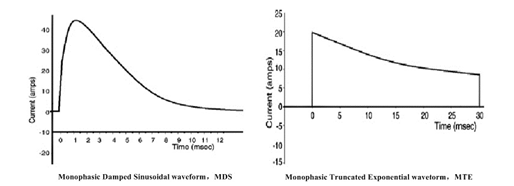
图 1
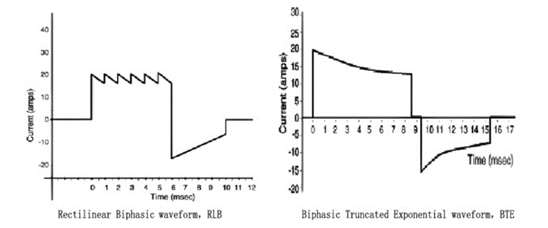
图 2
d)在所有的治疗模式下,注册申请人应提供阻抗测量的方法,应明确除颤波形对应的病人阻抗测量范围和精确度。
e)对于半自动和自动模式,注册申请人应描述心脏节律识别技术,陈述算法名称、算法原理和实现方式,明确心脏节律识别的性能指标,例如,分析时间、敏感度、特异性、真实预报价值和假阳性率等。
2.关键元器件及其质量控制要求
注册申请人应提供产品的工作原理图、产品总体设计方案和总体设计框图,应简述各单元模块的功能及相互关系,应明确手动体外除颤、半自动体外除颤、自动体外除颤相关的单元模块及其设计要求。
基于风险分析和管理、验证和确认等的工作,注册申请人应确定下述模块(包括但不限于)相关器件是否为关键元器件,并明确关键元器件的名称、型号、规格、制造商以及需要控制的规格参数和性能指标:
a)电源模块,例如,电源适配器、电池、电源模块等。
b)充电模块,例如,充电电容、充电高压管、充电变压器等。
c)放电模块,例如,放电桥中电子开关等。
d)控制模块,例如,硬件电路的MCU、扩展资源、通信电路等。
e)测量模块,例如,病人阻抗传感器,信号采集和转化电路等。
f)治疗附件,例如,除颤手柄、一次性使用除颤电极片等。
(二)生物相容性评价研究
注册申请人应对预期与人体接触附件进行生物相容性评价,例如,除颤手柄、可重复使用除颤电极、一次性使用除颤电极片、胸内勺形电极以及其他配合使用的附件,应提供细胞毒性、致敏、刺激或皮内反应项目的生物相容性评价资料,对于灭菌包装附件,还应提供热原和细菌内毒素项目的评价资料。
宜参考的现行有效标准有:
GB/T 16886.1-2011 医疗器械生物学评价第1部分:风险管理过程中的评价与试验;
GB/T 16886.5-2003 医疗器械生物学评价第5部分:体外细胞毒性试验;
GB/T 16886.10-2005 医疗器械生物学评价第10部分:刺激与迟发型超敏反应试验;
GB/T 16886.12-2005 医疗器械生物学评价第12部分:样品制备与参照样品;
GB/T 16886.18-2011 医疗器械生物学评价第18部分:材料化学表征;
GB/T 14233.2-2005 医用输液、输血、注射器具检验方法第2部分:生物学试验方法。
注册申请人宜参考GB/T 16886.1-2011评定程序的框架并且在风险分析和管理的基础上,根据产品与人体接触性质、接触时间和接触周期,考虑生物学评价项目,并按照图3进行生物学评价。
生物相容性评价研究资料应当包括:
1.生物相容性评价的依据和方法;
2.产品所用材料的描述及与人体接触的性质;
3.实施或豁免生物学试验的理由和论证;
4.对于现有数据或试验结果的评价。
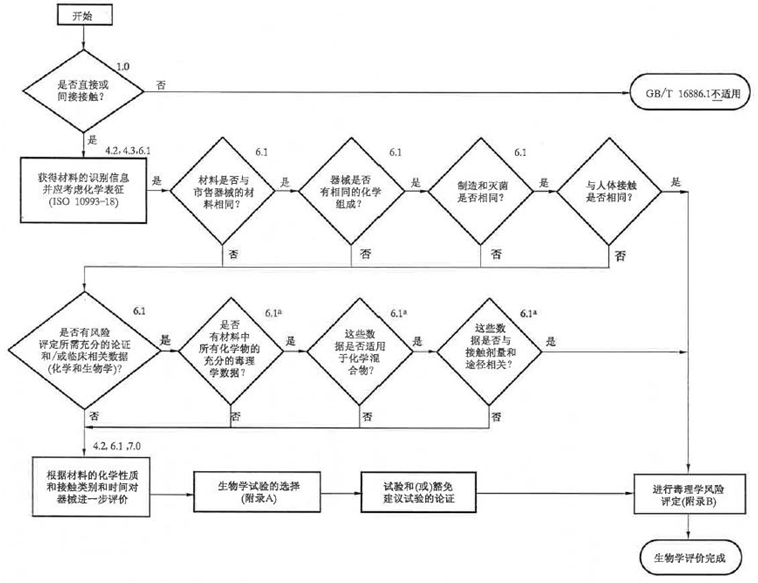
图 3
对于已获得注册批准上市的附件,在GB/T 16886.1-2011的4.7项中任一情况下,应重新评价材料或最终产品的生物相容性。
如果已获得注册批准上市的附件未发生GB/T 16886.1-2011中4.7项的情况,注册申请人应提供其法定制造商做出的未发生GB/T 16886.1-2011的4.7项所规定的重新评价情况的声明,并且注册申请人应提供该附件的医疗器械注册证复印件及其原生物学评价资料的复印件。
(三)产品有效期和包装研究
1.应明确电池和除颤电极片的有效期,并提供有效期的验证报告。
2.应明确申报产品的预期使用寿命,提供预期使用寿命的分析验证报告。
3.应提供可重复使用附件的使用次数验证资料,例如,可充电电池、除颤手柄、可重复使用除颤电极板、可重复使用除颤电极片等。
4.应提供在宣称的有效期内以及运输储存条件下,保持包装完整性的依据。
5.如产品为灭菌包装,应明确包装材料、灭菌工艺(方法和参数)、无菌保证水平和灭菌有效期,提供包装工艺确认报告、灭菌确认报告、灭菌效果验证报告和货架寿命验证报告。
(四)临床前动物实验
如必要,应提供体外除颤产品的动物实验研究资料,见本指导原则附录3。
(五)软件研究
体外除颤产品的软件一般属于软件组件,一般不宜单独注册。参考《医疗器械软件注册技术审查指导原则》的要求,应按照C级安全性级别来提供软件描述文档,应关注:
1.核心算法:
注册申请人应提供阻抗测量及补偿技术的算法名称、类型、用途和临床功能,如为全新算法,还应提供安全性和有效性的验证资料。
对于半自动和自动模式,注册申请人应提供心脏节律识别技术的算法名称、类型、用途和临床功能,如为全新算法,还应提供安全性和有效性的验证资料。
2.心脏节律识别功能的验证:
按照GB 9706.8-2009的6.8.3条款3)项,提供心律识别检测器的性能验证资料,包括心电数据库报告和性能验证报告。
a)心电数据库报告:
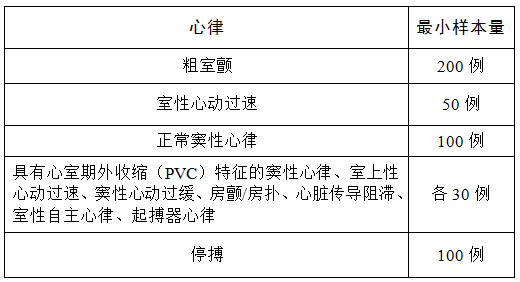
注册申请人应报告用于心律识别检测器性能验证的心电数据库,应描述记录方法、心律来源、心律选择基准,并且应提供评注方法和基准,例如,由具有多年心电图临床经验的专家组标注心电图数据。该数据库至少应满足表3的要求。
用于验证心脏节律识别的心电数据库应包括可除颤心律和非可除颤心律:
可除颤心律应包括不同幅度的粗室颤心律、不同频率和QRS波宽度的室性心动过速心律。
非可除颤心律应包括不同形态的QRS波数据(含干扰信号),并包括正常窦性心律、具有心室期外收缩(PVC)特征的窦性心律、室上性心动过速、窦性心动过缓、房颤、房扑、心脏传导阻滞、室性自主心律、起搏器心律、停搏。
表 3
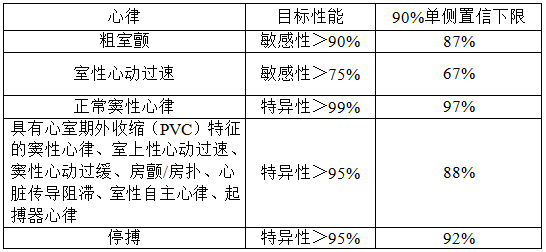
b)性能验证报告:
注册申请人应利用上述心电数据库验证心律识别检测器的性能并提供性能验证报告,验证结果应包括敏感性、特异性、真实预报价值和假阳性率,并且应满足表4要求。
表 4
五、临床评价资料
注册申请人应提供临床评价资料,可按照下述任一情形:
(一)通过同品种体外除颤产品临床数据进行的分析评价报告
按照本指导原则附录4和附录5,应通过分析评价同品种产品的临床数据来确认产品的安全性和有效性。
(二)临床试验资料
对于手动除颤、半自动或自动体外除颤产品,应按照本指导原则附录6来确认体外除颤治疗的安全性和有效性。
对于半自动或自动体外除颤产品,应按照本指导原则附录7来确认心脏节律识别器的安全性和有效性。
六、产品风险分析资料
注册申请人应提供体外除颤器的风险管理资料,见本指导原则附录1。
考虑到产品的临床使用场所(包括但不限于临床机构、户外/户内公共场所、交通工具中、患者家中等)和各种环境因素的相关风险[包括但不限于海拔、绝对湿度和相对湿度、热能(温度)、电磁环境、辐射环境、机械能(重力坠落和悬挂、碰撞、振动、运输)等],应在风险分析和管理过程中估计、评价并控制环境因素可能引起的相关风险,同时应说明提出风险控制措施的客观证据及验证确认资料,确认该产品在不同环境条件下的安全性、临床功能和性能。
七、产品技术要求
注册申请人应按照本指导原则附录2提供产品技术要求。
八、产品使用说明书
应明确产品的预期使用寿命,并明确预期使用寿命内产品的自检频率和要求、维护方法和维护频率。
九、注册单元的划分
1.技术原理相同但产品设计结构不同的产品原则上应划分为不同的注册单元。
2.治疗模式不同的产品应划分为不同的注册单元。例如,手动体外除颤器、半自动体外除颤器、自动体外除颤器、体外同步复律仪、体外除颤监护仪/系统应划分为不同的注册单元。
3.治疗波形不同的产品应划分为不同的注册单元,例如,双相波、单相波或其他波形等。
十、名词解释
体外电复律:在严重快速型心律失常时,利用外加的高能量脉冲电流通过心脏,使全部或大部分心肌细胞在瞬间同时除极,造成心脏短暂的电活动停止,然后由最高自律性的起搏点(通常为窦房结)重新主导心脏节律的治疗过程。
体外非同步电复律:在患者的QRS波和T波分辨不清或不存在时,不启用同步触发装置所进行的体外电复律。
体外除颤:在患者发生室颤(室扑或无脉室速)时进行的体外非同步电复律。
体外除颤器:通过电极将电脉冲施加在患者的皮肤或暴露的心脏,用来对心脏进行体外除颤的医用电气设备。

前—侧位:也叫前尖位或标准位,一个电极板放在右前壁锁骨下,靠近但不与胸骨重叠(注意,无论如何也不要将电极放在胸骨上,以免明显减弱除颤时放电时的能量;另一个电极板放在心尖(左乳头左侧,其中心位于腋中线上)。两块电极板之间的距离不应<10cm)。
前—后位:一个电极板放在左肩胛下区,另一个电极板放在胸骨左缘第四肋间水平。
前—左肩胛位:一个电极板放在右前壁锁骨下,另一个电极板放在背部左肩胛下。
前—右肩胛位:也叫尖后位,一个电极板放在心尖部,另一个电极板放在病人背后右肩胛角(注意,应避开脊柱)。
随机文件:随设备或附件所附带的文件,其内容包括对设备的使用者、操作者、安装者或装配者来说是全部重要的资料,特别是有关安全的资料。
生命支持设备或系统:至少包括一种预期有效地保持患者生命或复苏功能的设备或系统,且一旦该功能无法满足要求就很可能导致患者严重的伤害或死亡。
预期使用寿命:由制造商规定的电气设备或电气系统期望保持安全使用的时间(即保证基本安全和基本性能)。
注:参见IEC 60601-1: 2012的3.28项。
心律识别检测器:分析心电图并识别一个心脏节律是否为可电击的仪器。在半自动体外除颤器、自动体外除颤器中,其算法设计为保证心律失常检测的敏感性和特异性,以便在临床中对其进行除颤。
可除颤心律:若不及时地除颤,可能造成患者死亡的致死性心律。
非可除颤心律:不可电击的良性心律(甚至正常心律),特别是尚有脉搏的病人,因为除颤不能给这些病人带来任何益处,反而可能造成病情恶化。
除颤成功:终止心室颤动,除颤后5秒钟心电图显示心搏停止或非室颤无电活动。

十一、参考文献
(一)《医疗器械监督管理条例》(国务院令第650号)
(二)《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号)
(三)《医疗器械临床试验质量管理规范》(国家食品药品监督管理总局 中华人民共和国国家卫生和计划生育委员会令第25号)
(四)《医疗器械通用名称命名规则》(国家食品药品监督管理总局令第19号)
(五)GB 9706.1-2007 医用电气设备第1部分:安全通用要求
(六)GB 9706.8-2009 医用电气设备第2-4部分:心脏除颤器安全专用要求
(七)GB 18279-2000 医疗器械环氧乙烷灭菌确认和常规控制
(八)GB 18280-2000 医疗保健产品灭菌确认和常规控制要求辐照灭菌
(九) GB/T 14710-2009 医用电器环境要求及试验方法
(十)YY 0505-2012 医用电气设备第1-2部分:安全通用要求并列标准:电磁兼容要求和试验
(十一)YY 0670-2008 无创自动测量血压计
(十二)YY/T 0287-2003 医疗器械质量管理体系用于法规的要求
(十三)YY/T 0316-2008 医疗器械风险管理对医疗器械的应用
(十四)《医疗器械注册申报资料要求和批准证明文件格式》(国家食品药品监督管理总局公告2014年第43号)
(十五)《食品药品监管总局关于执行医疗器械和体外诊断试剂注册管理办法有关问题的通知》(食药监械管〔2015〕247号)
(十六)《医疗器械临床评价技术指导原则》(国家食品药品监督管理总局通告2015年第14号)
(十七)《药物临床试验的生物统计学指导原则》(国家食品药品监督管理总局通告2016年第93号)
(十八)《医疗器械产品技术要求编写指导原则》(国家食品药品监督管理总局通告2014年第9号)
(十九)《医疗器械软件注册技术审查指导原则》(国家食品药品监督管理总局通告2015年第50号)
(二十)《医疗器械生物学评价和审查指南》(国食药监械〔2007〕345号)
(二十一)IEC 60601-1:2012Medical electrical equipment - Part 1:General requirements for basic safetyand essential performance
(二十二)EN 1789:2007+A1: 2010Medical vehicles and their equipment - Road ambulances
(二十三)EN 13718-1: 2008 Medical vehicles and their equipment - Air ambulances - Part 1:Requirements for medical devices used in air ambulances
(二十四)Richard E. Kerber, MD, Chair; Lance B. Becker.MD; Joseph D. Bourland EE, PhD; Richard O. Cummins, MD. MPH; Alfred P. Hal lstrom,PhD; Mary B. Michos, RN;Graham Nichol, MD; Joseph P. Ornato, MD; William H. Thies, PhD; RogerD. White, MD; Bram D. Zuckerman, MD.Automatic External Defibrillators for Public Access Defibrillation: Recommendations for Specifying and Reporting Arrhythmia Analysis Algorithm Performance, Incorporating New Waveforms, and Enhancing Safety.AHA Scientific Statement
(二十五)CCTS工作组,贺佳(执笔)。临床试验中样本量确定的统计学考虑,中国卫生统计,2015,32(4)
(二十六)CCTS工作组,贺佳(执笔)。临床试验统计分析计划及统计分析报告的考虑。中国卫生统计,2015,32(3)
(二十七)杨兴华,詹思延。临床研究证据分级及评价。中国循证心血管医学杂志,2009,1(4)
(二十八)杨学宁,吴一龙。临床证据水平分级和推荐级别。循证医学,2003,3(2):111-113
(二十九)黄惠芳,朱莹,顾国浩。体外电除颤对心肌损伤标志物影响的实验研究。苏州大学学报(医学版)2009,29(5)
(三十)张晓东,何庆。双/单相波电击除颤治疗院前室颤的系统评价。西南军医,2009,11(4)
附录:1.风险管理资料
2.产品技术要求模板
3.动物实验研究
4.通过同品种体外除颤产品临床数据进行的分析评价报告
5.除颤波形及其性能参数的差异性研究
6.体外除颤治疗的临床试验研究
7.心脏节律识别器的临床试验研究

欢迎关注《医疗装备》官方公众号

